Modeling of Protein-Small Molecule Complexes
The hetero compound that I am studying is phenylalanine. I navigated to the RCSB protein data bank site and conducted a search for phenylalanine. From the list of results I received I chose to study L-Phenylalanine Dehydrogenase. The PDB identification for this protein is 1C1D and the hetero compound code Phe 361.
Once I downloaded the information for the protein, I opened the file in DS Visualizer (DSV). Then I opened a hierarchy window to asses the structure. I deleted all water molecules and a second amino acid chain that did not have my hetero compound embedded in the complex. I found my hetero compound under the “chain” category in the hierarchy window. I selected the hetero compound and copied and pasted it into another DSV window thereby “extracting” the amino acid. After extracting the compound, I had to adjust the amino acid to reflect its zwitterionic conformation. I then saved this file as an MDL.mol. After the compound was saved, I converted it into a ball and stick representation saved it as an .msv and .jpg file. The ball and stick representation of the “extracted” compound can be seen in the jpeg below.
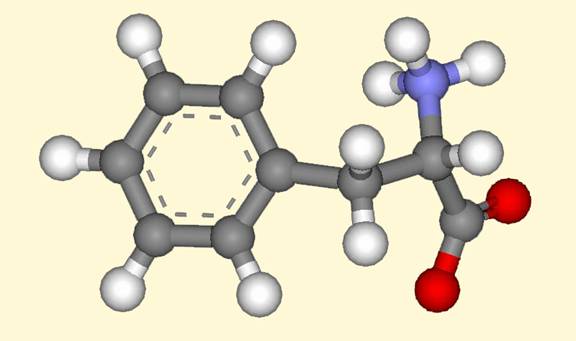
Figure 1 DS Visualizer image of Phe 361 obtained form the RCSB Protein Data Bank.
After creating the jpg file, I opened ChemSketch to draw the Lewis structure of my compound as observed at its physiological pH. I used the ball and stick picture to create the same compound with an atom modeling kit to see the stereo chemistry. I saved the file as a gif file and inserted it below.
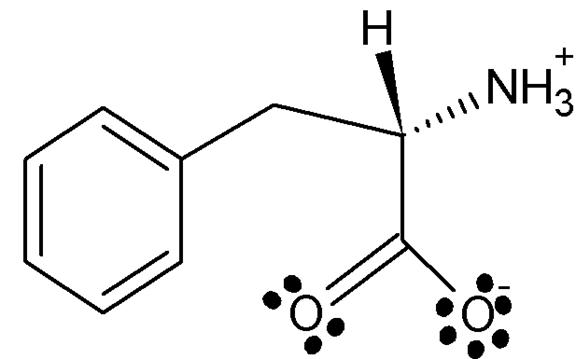
Figure 2 ChemSketch Lewis structure of Phe 361
Once the above was completed, I created a new web page entitled Protein Modeling and placed it into a directory named Protein that is located in my Projects directory. I then linked “Protein Modeling” to my CHEM 350 projects page. All files created were placed in the newly created protein directory and the images were placed in the images directory. I created a hyperlink to this word document and inserted the image of my extracted compound onto the Protein Modeling Page.
In part II I tailored the PDB file to clearly display the hetero compound phenylalanine. I opened the PDB file once again in DS Visualizer. I selected my hetero compound, and on the tool bar, clicked the “Display Style” button. Then under the atom tab I selected the “CPK” display, clicked apply and the hetero compound was displayed in a space filled representation. I then selected “A” in the hierarchy window and using the same steps above, brought up the “Display Style” menu once again. Under the atom tab I selected “None” and under the protein tab selected the “Solid ribbon” selection. I applied the changes and clicked ok. I then saved the file as an msv and jpeg file and added the image seen below.
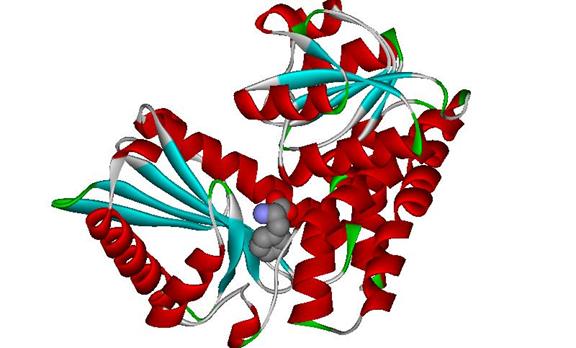
Figure 3 DS Visualizer image of Phe 361with L-Phenylalanine Dehydrogenase. PDB 1C1D obtained from RCSB Protein Data Bank
In part III of the assignment I opened the saved .mol format of my hetero compound in Chem 3D. I preformed a single point energy calculation by selecting the calculations drop down menu, highlighting the MM2 option and selecting Compute Properties. A new window opened up and under the Properties tab I choose Steric Energy Summary and clicked run. Once the single point energy calculation was completed I copied and pasted the data into the embedded excel spread sheet that makes up figure 4.
I saved the file in the default format then preformed an energy minimization calculation. The data was collected again by opening up the calculations dropdown menu, highlighting the MM2 option but this time choosing “minimize energy” as the job type. This brought up a new window and after ensuring that “Display Every Iteration” was selected and the “Minimum RMS Gradient” was set at 0.011, I clicked run. Once the test was completed, I again copied and pasted the data into the excel spread sheet found below. I also saved the energy minimized conformation of Phe 361 under a new name.
After the two tests were completed, I copied and pasted the two conformations found below.

Table 1 Comparison of the energy levels in the extracted form of Phe 361 and an energy minimized form of Phe. Energy levels in Kcal/mol.
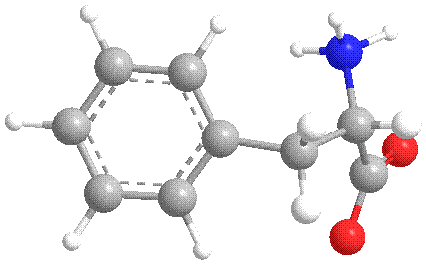
Figure 4 Extracted conformation of Phe 361
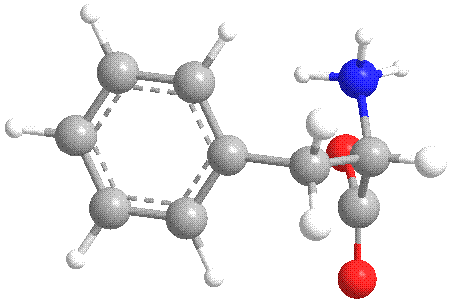
Figure 5 Energy minimized conformation of Phe.
Comparing the energies produced from the two conformations and looking at the structures of those conformations, it is possible to explain the differences in energies that are produced. Looking at the bottom line of the spread sheet you will see a total energy value and notice that this value is negative. This is because the more negative the energy, the more stable that conformation is. This is a law know as Gibbs free energy. I will now examine each value and try to account for the energy differences.
The “Stretch” value represents the energy associated with distorting bonds from their optimal length and has a difference in energy of 27.8248 Kcal/mol. The stretching difference that I noticed between the two conformations is between the carbon of the carboxylic acid and the carboxylate oxygen. In the extracted conformation the bond length is 1.253 Angstroms, and in the energy minimized conformation, this value is 1.480 Angstroms. The “Stretch” value is the second largest energy difference between the two conformations.
The “Bend” value is the energy associated with deforming bond angles from their optimal values. The difference between the two conformations occurs at the angle between the alpha carbon and the carbon that attaches the phenol ring. In the extracted conformation this angle is 120 degrees, and in the energy minimized conformation, the angle is 114 degrees. The difference in energy seems to favor the extracted conformation probably because the atoms are more spread out with the wider angle which would reduce steric strain. This is an interesting effect because the normal bond angle in a tetrahedan carbon is 109 degrees. Perhaps the widened angle is from other amino acids causing the bond angle to stretch.
The “Stretch-Bend” is the energy required to stretch the two bonds involved in a bond angle when that bond angle is severely compressed. This value did not make a large impact on the energy level with a difference of 0.0657 Kcal/mol.
The next value is the “Torsion” value which represents the energy associated with deforming torsional angles in the molecule from their ideal values. As in the “Stretch-Bend” value, this did not impact the over all energy levels between the two conformations. The difference between the two values is 0.0293 Kcal/mol with the energy minimized conformation having the smaller value.
The next two interactions are the van der Waals interactions (VDW). The non-1,4 VDW is the energy for the through-space interaction between pairs of atoms that are separated by more than three atoms. The non-1,4 VDW values were minimal with a difference of 0.1948 Kcal/mol, favoring the energy minimized conformation. The 1,4 VDW values represent the energy for the through-space interaction of atoms separated by two atoms. The difference between the two is larger than the non-1,4 VDW is a difference of -5.487 Kcal/mol. Oddly the extracted conformation is more energy favorable. This may be due to the interactions between the nitrogen group and the oxygen atoms of the carboxylic acid group. In the extracted conformation, it is the carbonyl oxygen that is closest to the nitrogen group. However, in the energy minimized conformation it is the carboxylate oxygen that is closest to the nitrogen group.
The next value is the “Charge/Charge” interaction which is explained by the attraction or repulsion of the charges on the atoms. This gives the biggest energy difference between the two conformations with a value of 41.2639 Kcal/mol. The energy difference favors the energy minimized conformation because the carboxylate oxygen has a more negative charge and is strongly attracted to the positive nitrogen group. Although in the extracted conformation of Phe 361 has the same interaction, it is the slightly less carbonyl oxygen that is interacting with the nitrogen group.
The “Charge/Dipole” interaction causes a minimal difference in energy between the two conformations with a value of –0.618 Kcal/mol favoring the extracted molecule. The difference in energy could be caused by which oxygen atom the nitrogen group interacts with.
The “Dipole/Dipole” charge is the energy associated with the interaction of bond dipoles. The energy involved most likely comes from the same group of atoms that causes the “Charge/Dipole” and “Charge/Charge” energy levels, the nitrogen group interacting with either of the two oxygen atoms of the carboxylic group. The difference in energy is 0.0618 Kcal/mol, favoring the energy minimized conformation.
The last value is the “Total,” this is the sum of all of the listed energies. The extracted conformation gives a value of -25.3 Kcal/mol and the energy minimized value is -86.7421 Kcal/mol. The difference between the two is 61.4421 Kcal/mol favoring the energy minimized conformation.
In summary, the “Charge/Charge” interaction made the greatest difference between the two conformations. This is due to the interaction of the nitrogen group with the more negative carboxylate oxygen or the nitrogen group with the carbonyl oxygen. The interaction that least affected the total is the “Torsion” value. The difference between the two was the smallest out of all of the calculated values.
To illustrate the conformational differences between the two compounds, an overlaying jpg image was created and inserted below. To create the image, I opened the extracted and energy minimized conformations in Chem3D. I copied the extracted structure and pasted it into the energy minimized screen. Once I had the two conformations in the same Chem3D window, I saved the file in Chem 3D’s default format.
Next I opened a new panel called “Fragments” by choosing “View” in the drop down menu, then choosing “Model Explorer.” I selected one of the fragments from the plane which highlighted the corresponding fragment in the model. Once the atoms were highlighted, from the menu bar I chose “Structure,” “Overlay,” and “Set Target.” I then chose the other fragment and in the same “Structure” menu, I again chose “Overlay,” but this time chose the “Fast Overlay” option which gave me the image below. I rotated the structure to give the best representation of the differences between the two conformations. To “clean up” the structure, from the menu bar I chose “View,” “Model Display,” “Show Hydrogen Atoms” then I chose “Show Polar,” which hid all hydrogen atoms except those attached to the nitrogen atom.
Examining the structure it seems that the pieces that fit together the best are the upper two carbons on the phenyl ring and the nitrogen atoms. The rest of the atoms are clearly very contorted with the oxygen atoms of the carboxyl group being affected the most. Looking at the oxygen atoms closely, I noticed that the oxygen atom closest to the nitrogen atom flipped. This observation was used to explain some of the energy differences in the values of figure 4. The oxygen atoms being flipped between the two conformations causes the bonds to go off in different angles causing the biggest difference between the two structures.
Afterwards, I updated my molecular modeling document along with the hyper link on the web site I created. I also inserted a link to view the overlay image in the jpg format on the web site.
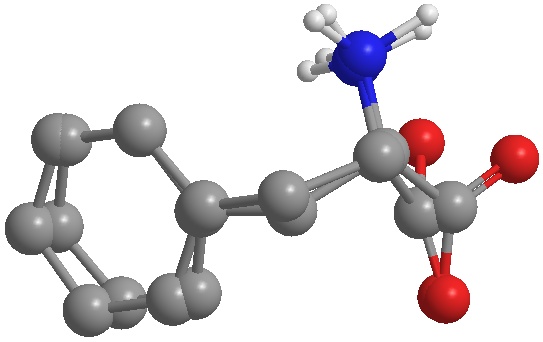
Figure 6 Overlaid view of the extracted Phe 361 conformation and the energy minimized conformation of Phe.
In part IV of the assignment, I had to display the interactions between my hetero compound and the amino acids that act upon it. To see which residues acted upon my compound, I went to the PDB Sum website and entered in my protein’s 4 digit code that I obtained from the PDB website. Once my protein came up I clicked on the “Protein” tab on the top navigation and the wiring diagram shown below came up
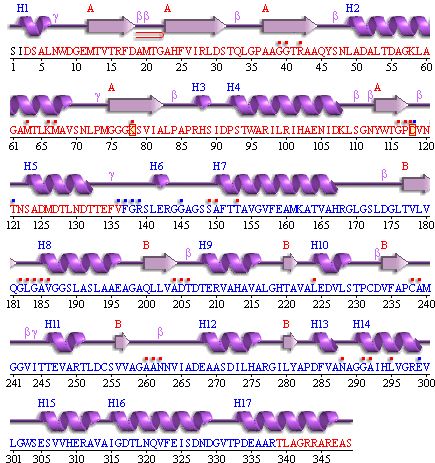
Figure 7 wire diagram of L-Phenylalanine Dehydrogenase obtained from the PDB Sum website.
. After I downloaded the wire diagram, I clicked on the “Ligands” tab, and then I selected Phe 361 on the left navigation. This brought up the “Ligplot” for my hetero compound. For a better picture of the “Ligplot,” I downloaded the PDF file and inserted it as figure 9 found below.
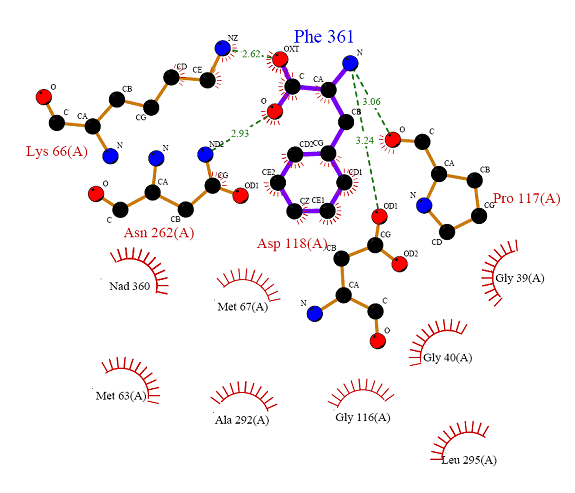
Figure 8 Ligplot display of Phe 361 and interacting amino acid residues.
Now that I could tell which amino acid residues act on Phe 361, I opened my protein file in DSV and selected “Chain A” in the hierarchy window. Opening the “Display Style” window by typing in “Ctrl + D” I changed the protein color to grey. I then saved the presentation of the protein in the msv and jpg formats and inserted the image below.
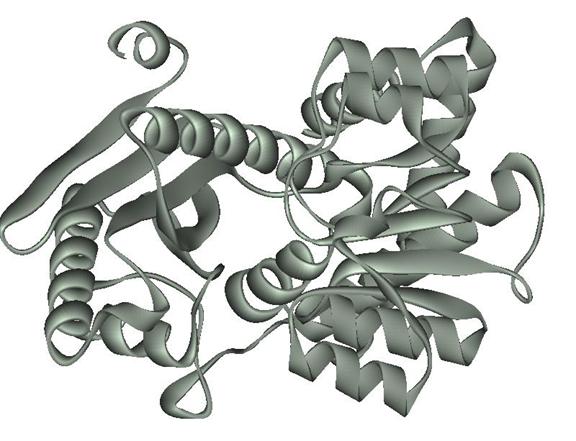
Figure 9 Protein L-Phenylalanine Dehydrogenase represented in solid ribbon colored grey without the hetero compound phenylalanine.
Next I found my hetero compound in the hierarchy window, selected it, and brought up the “Display Style” window again and chose to display Phe 361 in a ball and stick format. Then, with the “Ligplot” as an aid, I selected the amino acid residues that interact with my hetero compound.
Once the residues were selected, I
right clicked with the mouse and selected “Display Style” once again. In the
“Atom” tab, I chose to display them as stick structures and I also changed the
color to a pale yellow. Once that was done I right clicked the mouse again and
this time selected “Label.” In this window I set the “Object” to amino acid
and changed the font to courier. To see the effect of what I did, I selected
the “A” chain and again went into the “Display Style” window and in the
“Protein Tab” turned off the “Display Style” so that the only thing showing was
the hetero compound and the interacting amino acids.
I next wanted to display the hydrogen bonds that occur. I chose “Structure,” “Monitor,” and “Hydrogen bond,” which displayed the green dashed lines in figure 9. I noticed that some of the bonds that were displayed on the “Ligplot” were not displayed on the DSV. To display the missing bonds, I selected “HBond Monitor” in the hierarchy window, in the display window; I right clicked the mouse and selected “Attributes of HBond Monitor.” Once the new window came up I changed the “Distance Criterion” to 3.24, the greatest distance of a hydrogen bond according to the “Ligplot.”
Once the hetero compound and interacting amino acids were clearly displayed, I rotated the structure to represent what was on the “Ligplot” keeping the amino acids that were in the form of “Eye Lashes” above or behind the hetero compound. I saved this view as an msv and jpg file.
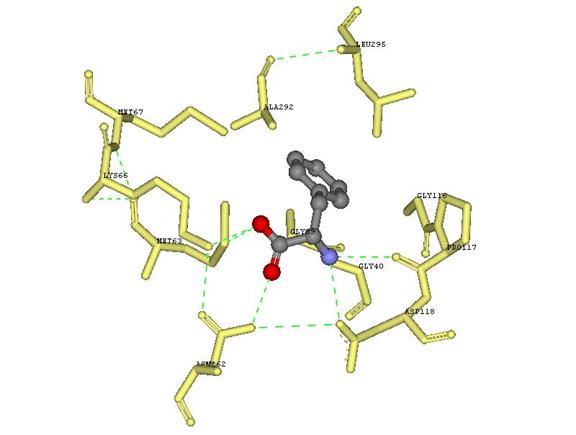
Figure 10 Phe 361 with interacting amino acid residues displayed to mimic the Ligplot obtained from the PDB Sum website.
To visualize how the active site would look, I turned the protein display back on set to solid ribbon and rotated the protein to give the best view. I saved the files as a msv and jpg file and inserted the image below.
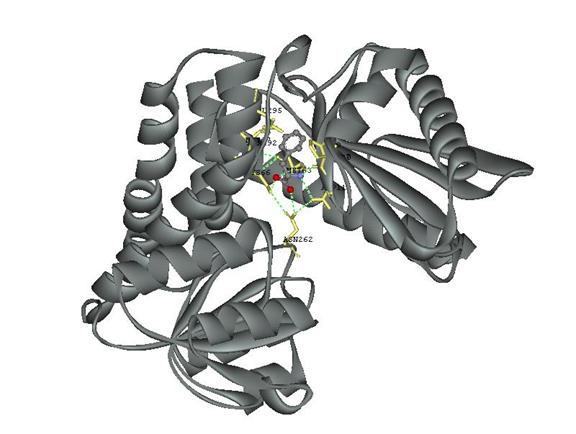
Figure 11 Display of L-Phenylalanine Dehydrogenase with the active site displayed in the stick format in yellow and the hetero compound in ball and stick with the atoms in their normal color.
In part V of the assignment, I will talk about the protein-hetero compound interactions as seen in the Ligplot figure above and use DSV to aid me in explaining these interactions. HBA denotes a hydrogen bond acceptor, and HBD is a hydrogen bone donor.
Note: Looking at the Ligplot and observing the interactions in DSV, I noticed that the amino acids that were represented as eyelashes on the Ligplot seemed to help stabilize the amino acids that directly interacted with Phe 361 by hydrogen bonds.
|
Table 1 Protein Ligand Interactions
|
|||
|
Amino Acid Residue |
Hetero compound atom(s) that interact with the amino acid residue |
Type of Interaction |
|
|
Residue Number |
Reacting Atoms |
||
|
Gly 39, 40 |
??? |
???? |
The two Gly molecules don’t seem to be interacting with anything. |
|
Met 63 |
Back bone carboxylate. |
None |
Met 63’s carboxylate is a HBA to two other amino acid residues. One from the back bone amino group of Met 67 and the other from the backbone amino group of Lys 66, both of which are HBDs. |
|
Lys 66 |
Side chain amino group and amino group on back bone. |
Carboxylate |
The side chain amino group is a HBD to the HBA carboxylate of the hetero compound. The back bone amino group is also a HBD to the HBA carboxylate of Met 63. |
|
Met 67 |
Back bone amino group. |
None |
The amino group is a HBD to the HBA carboxylate of Met 63. |
|
Gly 116 |
??? |
None |
The Gyl molecule does not seem to be interacting with anything. |
|
Pro 117 |
Back bone carboxylate. |
NH3 |
The carboxylate acts as a HBA to the HBD amino group attached to the hetero compound. |
|
Amino Acid Residue |
Hetero compound atom(s) that interact with the amino acid residue |
Type of Interaction |
|
|
Residue Number |
Reacting Atoms |
||
|
Asp 118 |
Side chain carboxylate. |
NH3 |
The carboxylate acts as a HBA to the HBD amino group attached to the hetero compound. The carboxylate also acts as a HBA to the HBD amide nitrogen of Asn 62 |
|
Asn 262 |
Side chain amide group. |
Carboxylate |
The amide nitrogen acts as a HBD to the HBA carboxylate of the hetero compound. The amide oxygen also acts a HBA to the HBD amino side chain of Lys 66. |
|
Ala 292 |
Back bone amino group and carboxylate. |
Carboxylate |
The amino group is a HBD to the HBA carboxylate of Phe 361. The carboxylate of Ala 292 is a HBA from the HBD amino group on the side chain of Leu 295. |
|
Leu 295 |
Back bone amino group. |
None |
The amino group is a HBD to the HBA carboxylate of Ala 292. |
References
Brunhuber, N.M., Thoden, J.B., Blanchard, J.S., Vanhooke, J.L. Biochemistry, 2000, 39, 9174-87
http://mutex.gmu.edu:2314/cgi-bin/article.cgi/bichaw/2000/39/i31/pdf/bi000494c.pdf.
PDB ID: 1C1D
Brunhuber, N.M., Thoden, J.B., Blanchard, J.S., Vanhooke, J.L.
Rhodococcus L-phenylalanine dehydrogenase: kinetics, mechanism, and structural basis for catalytic specificity.
Biochemistry 39 pp. 9174-87 (2000). http://www.pdb.org/pdb/explore/explore.do?structureId=1C1D.
PDB Sum 1C1D. Retrieved April 26, 2008 at
http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl
Slayden, S. Modeling of Protein-Small Molecule Complexes. Retrieved 4/24/08. http://classweb.gmu.edu/sslayden/Chem350/assignments/molmod/protein-modeling.htm